Manufacturers of combination products should seek regulatory advice and operational support from the expert Celegence team to ensure that essential documentation and clinical evidence for your product portfolio complies under the new MDR. To discuss how our team can strategically partner with yours, reach out to us at [email protected], contact us online or read more about Celegence’s medical device services.
Drug Device Combinations (DDCs) have become more and more prevalent as a method of administration for medicinal products. Over the past several years, there has been a substantial increase in the number of scientific advice requests and marketing authorization applications for products that incorporate drug and device components, or drug-device combinations. In essence, it reflects the potential for devices to decrease the burden on patients and providers. Notably, this includes products with automated functions that help patients manage self-administration of medicines.
Drug Device Combinations such as pre-filled syringes or pre-filled pens are considered to “have the potential to make treatments safer, more effective, or more convenient or acceptable to patients.” However, they must fulfill a significant set of additional requirements. The US and EU have profoundly different systems for determining assessment routes for drug (or biologic) and device combinations.
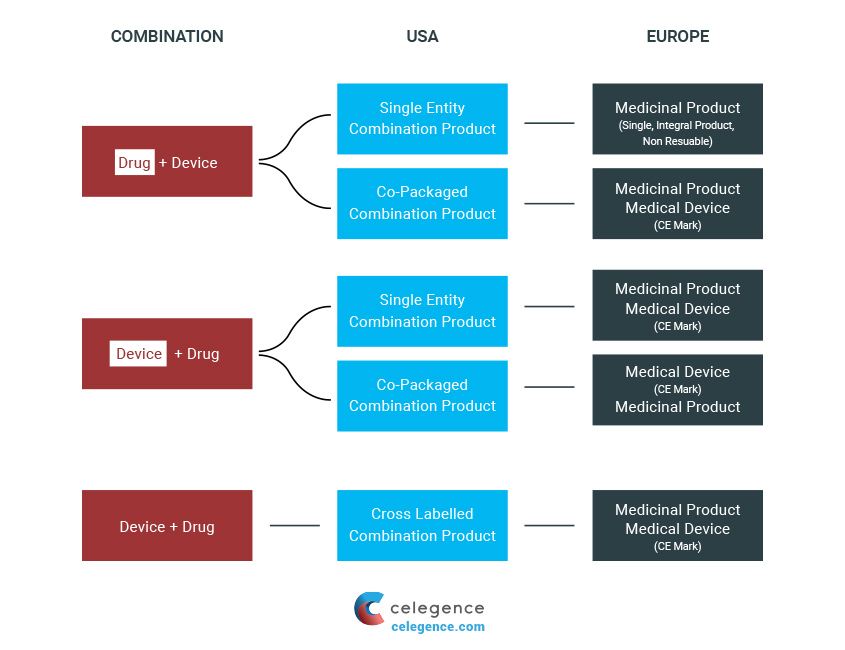
Combination Products Definitions
In the US, these products are termed as “combination products,” and the lead division is selected based on the primary mode of action. For relevant aspects of the product, other divisions are chosen. However, in the EU, the process is slightly different as the term “combination product” is not recognized officially. Although the products are still assessed based on the primary mode of action, there is also the determination of two primary assessment formats: either medicinal product or medical device.
The EU regulates DDC Products as a medicinal product or as a medical device. Hence, there is no consistent definition given in the EU for such products. The European Medicines Agency (EMA) provides the most meaningful definition describing the combination and is stated as:
“The medical device may be supplied as an integral component of the medicinal product (e.g. pre-filled syringe, auto-injector), or separately (copackaged; e.g. oral syringe, pen-injector), as a non-integral combination with the medicinal product, or independently marketed (in cases where the device meets the requirements for the necessary delivery system stated in the Summary of Product Characteristics (SmPC) for the medicinal product).”
A single integral DDC is described as follows:
“If a medical device used to administer a medicinal product is placed on the market in such a way that the device and medicinal product form a single integral product which is intended exclusively for use in the given combination and which is not reusable, that single integral product shall be governed by Directive 2001/83/EC or Regulation (EC) No 726/2004, as applicable.”
The Existing System for CE Marking Combination Products
The Medicinal Product Directive (MPD) mandates evidence of CE marking but does not detail the requirements for non-CE marked devices. According to Article 1 Subpart 3 of the MPD, devices in which ‘the device and medicinal product form a single integral product which is intended exclusively for use in the given combination and which is not reusable’ were governed by the MPD. Additionally, the essential requirements of Annex I in the MDD was applicable to establish safety and performance of the device.

The New System for CE Marking Combination Products – Article 117
The EMA, which oversees the European Union’s pharmaceutical market, has begun rolling out guidance and clarifications regarding Medical Devices Regulation (MDR) compliance issues for manufacturers of combination products with drug and medical device components. As per the EMA guideline, manufacturers of drug-device combination products will have to meet MDR Article 117 regulatory requirements. The EMA notes that Article 117 of the Regulation requires Notified Body (NB) involvement for European market authorization of a medicinal product that incorporates an integral medical device, or drug-device combination product.
Article 117 of the new EU MDR amends Annex I of the Medicinal Product Directive (MPD) 2001/83/EC, point 12 of Section 3.2. This states that:
“(12) Where, a product is governed by this Directive, the marketing authorization dossier shall include, where available, the results of the assessment of the conformity of the device part with the relevant general safety and performance requirements set out in Annex I to that Regulation contained in the manufacturer’s EU declaration of conformity or the relevant certificate issued by a notified body allowing the manufacturer to affix a CE marking to the medical device.
If the dossier does not include the results of the conformity assessment referred to in the first subparagraph and where for the conformity assessment of the device, if used separately, the involvement of a notified body is required in accordance with Regulation (EU) 2017/745, the authority shall require the applicant to provide an opinion on the conformity of the device part with the relevant general safety and performance requirements set out in Annex I to that Regulation issued by a notified body designated in accordance with that Regulation for the type of device in question.”
The change mainly applies to integrated, non-reusable products where the drug element is the primary mode of action. Fundamentally, the device element of a medicinal product – when integral (classified either as Class I [i.e. sterile], Class IIa, Class IIb or Class III), non-reusable and intended exclusively for use in the given combination – needs to conform to the Annex I (MDR) GSPR and without the requirement to be regulated as a CE-medical device. Integral devices classified as Class I devices (i.e. non-sterile) are not subject to an NB opinion.
Although a short article, it introduces a new requirement for NB involvement in medicinal products with an integral medical device. Marketing such products as a ‘medicinal product’ would require manufacturers to seek a Notified Body Opinion (NBOp). The NB will access and confirm if the device is compliant with the relevant General Safety and Performance Requirements (GSPR) and issues an NBOp report to the manufacturer. The NBO report is expected to be submitted to the EMA in the Market Authorisation Application (MAA) by the manufacturer. This will be assessed by the Competent Authority and the final approval of the product will remain with the EMA.
Amendment of the MPD and its Impact on the MAA
With the changes implemented by the EU MDR, the MAA must include either a:
- CE certificate issued by a NB for the medical device component, or
- Notified Body Opinion (NBO) on the conformity of the device
Scenario 1 would require manufacturers of the medical devices to obtain CE marking with the aid of an appropriate conformity assessment process applicable to the device. The CE certificate would then need to be submitted to the MAA.
Scenario 2 where a CE mark has not been issued to the device component on its own, the manufacturer must reach out to an NB and seek their opinion on the conformity of the device, and provide the NBO report in the MAA.
Role of Notified Bodies in MMA Submission
- To provide an NBO, the NBs will require access to the Technical Documentation of the manufacturer that provides sufficient evidence/scientific data to support product claims.
- Applicable to initial MMA submissions as well as submissions of “substantial design changes to the device component” (including “addition or full replacement of the device”)
- Technical documentation expectations by the NB are detailed in the Annex II of the MDR.
- Information that the NB would require to support a conformity assessment for medical devices includes:
- Risk Management Documentation
- Safety and Performance Data including clinical data
- Functionality Testing (accuracy of dosage, performance of device)
- Instructions for Use/Training for Users
- Usability/Human Factor Testing
- Compatibility Testing
- Sterilization Validation
- Sterility Test Data
- Packaging Validation Testing

Challenges Faced by Notified Bodies
- With the increase in requirements of the MDR, the number of NBs and their scope will decrease
- The available number of NBs qualified to assess DDCs may be lower than expected and this can turn out to be challenging for pharmaceutical manufacturers
- The expectations regarding the presentation of data showing compliance with the GSPRs are not yet well defined
- The assessment report of the NB concluding the requested opinion should be designed in a way that the medicine’s competent authority can rely on it for the final marketing authorization (MA). Hence representatives of the NB and the EMA should discuss the expectations and requirements in advance to ensure a consistent procedure
Next Steps for Manufacturers to Implement Article 117
- Identify a NB intending to be EU MDR designated for your specific technology
- Assess which GSPRs are applicable to your products
- Obtain scientific data that demonstrates conformance to the applicable GSPRs
- Assemble Technical Documents to support the medical device components
Summary – MDR Article 117 and Implications for Drug-device Combination Products
- For manufacturers who intend to qualify drug-device combination products, it is vital that their marketing authorization dossiers include the results of the conformity assessment or the CE Mark issued by the NB for device components. Furthermore, for device components that lack CE Marking, manufacturers will have to submit opinions from NBs regarding conformity of those components to the MDR requirements.
- A product’s device and medicinal components must form a “single integral product”, the product must be intended for use only in the drug-device combination; and the product must not be reusable. Only if these parameters are applicable, the product will have to comply with the requirements of MDR Article 117.
- For new marketing authorization applicants to establish that the device component of a drug-device combination product meets the MDR requirements established in Annex I of the Regulation, will require a DoC or CE Mark. If not, they must provide opinions from NBs supporting conformity to MDR Annex I rules.